Functional Group Filter
Here is a problem I have come across recently:
Given a list of SMILES is there a tool or way of recognising which functional groups each SMILES has ?
Here is the list of functional groups that I would like to examine:
- Acid (carboxylate)
- Alcohol
- Alkene
- Anhydride
- Amide
- 1y Amide
- 2y Amide
- 3y Amide
- Amine
- 1y Amine
- 2y Amine
- 3y Amine
- Heteroaromatic Amines
- Anilines
- Nitrogen Bases
- Aromatic
- Carbonate
- Ester
- Ether
- Ketone/Aldehyde
- Halogen
- Chloro
- Fluoro
- Hydrocarbon
- Nitrile
- Nitro
- Phosphorus Containing
- Sulfur Containing
- Sulfide
- Sulfoxide
- Sulfite
- Sulfone
- Urea
- Silicone
- Other
RDKit offers some of this functionality through its rdkit.Chem.Fragments module. Although covering enough groups from the above collection, this is not a comprehensive list.
As always, if something is not available, I like to have a go at building it my self.
I ended up defning 37 functions - one for each of the above groups (and a couple more). I was only interested in whether a functional group is present or not, and not how many times a group might be present as well. As the code for each functional group filter is long, I will first demo the use, and provide the code at the end.
I hope you find this functionality useful and sensible. If you spot any errors or improvements, please contact me.
First let’s import the required modules:
# pandas and numpy are always a must
import pandas as pd
import numpy as np
# rdkit for sensible cheminformatics
from rdkit import Chem
from rdkit.Chem import Fragments
Let’s define a function that will be taking a pandas dataframe with a column containing SMILES and returning a table (dataframe) with functional group names (as columns) and whether each row entry has that functional group:
def ApplyFGroupsFunctions(data, smiles_col = 'SMILES'):
''' Takes a dataframe with a 'SMILES' column and returns a new dataframe
with functional groups columns containing 'Y' or 'N' values. '''
fgroup_cols = {"Alcohol": [AlcoholGroup(smi) for smi in data[smiles_col]],
"Alkene": [AlkeneGroup(smi) for smi in data[smiles_col]],
"Alkyne": [AlkyneGroup(smi) for smi in data[smiles_col]],
"Anhydride": [AnhydrideGroup(smi) for smi in data[smiles_col]],
"Aromatic": [AromaticGroup(smi) for smi in data[smiles_col]],
"Amide": [AmideGroup(smi) for smi in data[smiles_col]],
"Amine": [AmineGroup(smi) for smi in data[smiles_col]],
"Aniline": [AnilineGroup(smi) for smi in data[smiles_col]],
"BasicN": [BasicNGroup(smi) for smi in data[smiles_col]],
"Bromo": [BromoGroup(smi) for smi in data[smiles_col]],
"Carbonyl": [CarbonylGroup(smi) for smi in data[smiles_col]],
"Carbonate": [CarbonateGroup(smi) for smi in data[smiles_col]],
"Chloro": [ChloroGroup(smi) for smi in data[smiles_col]],
"Ester": [EsterGroup(smi) for smi in data[smiles_col]],
"Ether": [EtherGroup(smi) for smi in data[smiles_col]],
"Fluoro": [FluoroGroup(smi) for smi in data[smiles_col]],
"Halogen": [HalogenGroup(smi) for smi in data[smiles_col]],
"HetArAmine": [HetArAmineGroup(smi) for smi in data[smiles_col]],
"Hydrocarbon": [HydrocarbonGroup(smi) for smi in data[smiles_col]],
"Iodo": [IodoGroup(smi) for smi in data[smiles_col]],
"Ketone/Aldehude": [KetAlGroup(smi) for smi in data[smiles_col]],
"Nitrile": [NitrileGroup(smi) for smi in data[smiles_col]],
"Nitro": [NitroGroup(smi) for smi in data[smiles_col]],
"Phosporus": [PhosporusGroup(smi) for smi in data[smiles_col]],
"PrimaryAmide": [PrimaryAmideGroup(smi) for smi in data[smiles_col]],
"PrimaryAmine": [PrimaryAmineGroup(smi) for smi in data[smiles_col]],
"SecAmine": [SecAmineGroup(smi) for smi in data[smiles_col]],
"SecAmide": [SecAmideGroup (smi) for smi in data[smiles_col]],
"Silicone": [SiliconeGroup(smi) for smi in data[smiles_col]],
"Sulfide": [SulfideGroup(smi) for smi in data[smiles_col]],
"Sulfone": [SulfoneGroup(smi) for smi in data[smiles_col]],
"Sulfur": [SulfurGroup(smi) for smi in data[smiles_col]],
"Sulfite": [SulfiteGroup(smi) for smi in data[smiles_col]],
"Sulfoxide": [SulfoxideGroup(smi) for smi in data[smiles_col]],
"TertAmine": [TertAmineGroup(smi) for smi in data[smiles_col]],
"TertAmide": [TertAmideGroup(smi) for smi in data[smiles_col]],
"Urea": [UreaGroup(smi) for smi in data[smiles_col]]}
new_cols = pd.DataFrame(fgroup_cols)
return new_cols
Now let’s define a dataframe with a few molecules to test the above:
smiles = ['CC(C)=O', 'CC(=O)N(C)C', 'CC#N', 'CC(=O)OCC',
'CC(=O)OCC', 'C(=O)(OC(C)C)OC', 'C(=O)(NC)NC',
'O1CCN(C)CC1', 'C1=CC=CC2=C1C=CN=C2', 'C1=CC=CC2=C1C(=O)OC2=O',
'C1=CC=C(C4=C1C2CC(C)(N(C(N2C)=O)CCCC3=CC=C(C=C3)OC)O4)F',
'C1C(C(N1C2=CC=C(C(=C2)C(F)(F)F)C#N)=O)C(C)=O',
'CN1C[C@@H](C=C2[C@H]1CC3=CNC4=CC=CC2=C34)C(=O)O', 'CN(C)CCC1=CNC2=C1C(=CC=C2)OP(=O)(O)O']
names = ['Acetone', 'DMF', 'Acetonitrile', 'Ethyl Acetate',
'Isopropyl Acetate', 'Methyl Isopropyl Carbonate', 'Bis(Methyl) Urea',
'N-Methyl Morpholine', 'Isoquinoline', 'Aromatic Anhydride',
'Chromopynone Analogue', 'Lactam', 'Lysergic acid', 'Psylocybin']
data = pd.DataFrame(zip(names, smiles), columns = ['Names', 'SMILES'])
data
| Names | SMILES | |
|---|---|---|
| 0 | Acetone | CC(C)=O |
| 1 | DMF | CC(=O)N(C)C |
| 2 | Acetonitrile | CC#N |
| 3 | Ethyl Acetate | CC(=O)OCC |
| 4 | Isopropyl Acetate | CC(=O)OCC |
| 5 | Methyl Isopropyl Carbonate | C(=O)(OC(C)C)OC |
| 6 | Bis(Methyl) Urea | C(=O)(NC)NC |
| 7 | N-Methyl Morpholine | O1CCN(C)CC1 |
| 8 | Isoquinoline | C1=CC=CC2=C1C=CN=C2 |
| 9 | Aromatic Anhydride | C1=CC=CC2=C1C(=O)OC2=O |
| 10 | Chromopynone Analogue | C1=CC=C(C4=C1C2CC(C)(N(C(N2C)=O)CCCC3=CC=C(C=C... |
| 11 | Lactam | C1C(C(N1C2=CC=C(C(=C2)C(F)(F)F)C#N)=O)C(C)=O |
| 12 | Lysergic acid | CN1C[C@@H](C=C2[C@H]1CC3=CNC4=CC=CC2=C34)C(=O)O |
| 13 | Psylocybin | CN(C)CCC1=CNC2=C1C(=CC=C2)OP(=O)(O)O |
new_cols = ApplyFGroupsFunctions(data)
#stitch the data together
data = pd.concat([data, new_cols], axis=1)
#check that it worked
data.head()
| Names | SMILES | Alcohol | Alkene | Alkyne | Anhydride | Aromatic | Amide | Amine | Aniline | ... | SecAmide | Silicone | Sulfide | Sulfone | Sulfur | Sulfite | Sulfoxide | TertAmine | TertAmide | Urea | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | Acetone | CC(C)=O | N | N | N | N | N | N | N | N | ... | N | N | N | N | N | N | N | N | N | N |
| 1 | DMF | CC(=O)N(C)C | N | N | N | N | N | Y | N | N | ... | N | N | N | N | N | N | N | Y | Y | N |
| 2 | Acetonitrile | CC#N | N | N | N | N | N | N | N | N | ... | N | N | N | N | N | N | N | Y | N | N |
| 3 | Ethyl Acetate | CC(=O)OCC | N | N | N | N | N | N | N | N | ... | N | N | N | N | N | N | N | N | N | N |
| 4 | Isopropyl Acetate | CC(=O)OCC | N | N | N | N | N | N | N | N | ... | N | N | N | N | N | N | N | N | N | N |
5 rows × 39 columns
That is lovely!
Let’s see if we can use it to slice the data appropriately:
mask = data['Aromatic'] == 'Y'
selected = data[mask]
selected
| Names | SMILES | Alcohol | Alkene | Alkyne | Anhydride | Aromatic | Amide | Amine | Aniline | ... | SecAmide | Silicone | Sulfide | Sulfone | Sulfur | Sulfite | Sulfoxide | TertAmine | TertAmide | Urea | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 8 | Isoquinoline | C1=CC=CC2=C1C=CN=C2 | N | N | N | N | Y | N | Y | N | ... | N | N | N | N | N | N | N | Y | N | N |
| 9 | Aromatic Anhydride | C1=CC=CC2=C1C(=O)OC2=O | N | N | N | Y | Y | N | N | N | ... | N | N | N | N | N | N | N | N | N | N |
| 10 | Chromopynone Analogue | C1=CC=C(C4=C1C2CC(C)(N(C(N2C)=O)CCCC3=CC=C(C=C... | N | N | N | N | Y | Y | N | N | ... | N | N | N | N | N | N | N | Y | Y | Y |
| 11 | Lactam | C1C(C(N1C2=CC=C(C(=C2)C(F)(F)F)C#N)=O)C(C)=O | N | N | N | N | Y | Y | N | Y | ... | N | N | N | N | N | N | N | Y | N | N |
| 12 | Lysergic acid | CN1C[C@@H](C=C2[C@H]1CC3=CNC4=CC=CC2=C34)C(=O)O | N | Y | N | N | Y | N | Y | N | ... | N | N | N | N | N | N | N | Y | N | N |
| 13 | Psylocybin | CN(C)CCC1=CNC2=C1C(=CC=C2)OP(=O)(O)O | N | N | N | N | Y | N | Y | N | ... | N | N | N | N | N | N | N | Y | N | N |
6 rows × 39 columns
Let’s check visualise these molecules to show that they have an aromatic group:
legend = [name for name in selected['Names']]
mols = [Chem.MolFromSmiles(smi) for smi in selected['SMILES']]
img = Chem.Draw.MolsToGridImage(mols=mols, legends=legend)
img
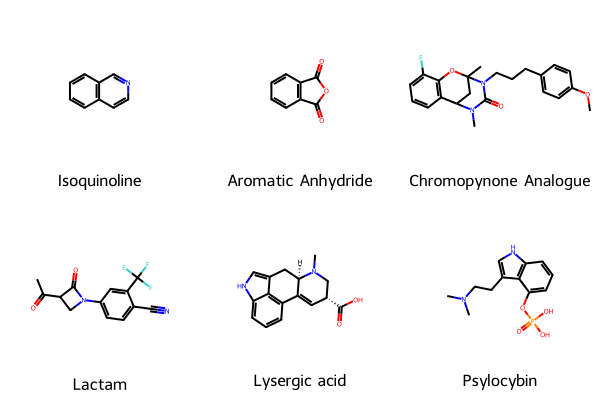
That’s nice to see.
I hope you find this functionality useful and sensible. If you spot any errors or improvements, please contact me.
As always the code can also be found on Github here.
Here is the code for the Functional Group Recognition Functions:
Functional_Groups_Functions
### --- DEFINE FUNCTIONAL GROUP RECOGNITION FUNCTIONS ---
def AlcoholGroup(smi):
mol = Chem.MolFromSmiles(smi)
OH_groups = 0
OH_groups += Chem.Fragments.fr_Al_OH(mol)
OH_groups += Chem.Fragments.fr_Ar_OH(mol)
if OH_groups > 0:
return "Y"
else:
return "N"
def AmideGroup(smi):
mol = Chem.MolFromSmiles(smi)
amide_groups = 0
amide_groups += Chem.Fragments.fr_amide(mol)
if amide_groups > 0:
return "Y"
else:
return "N"
def PrimaryAmideGroup(smi):
mol = Chem.MolFromSmiles(smi)
pri_amide_groups = 0
pri_amide_groups += Chem.Fragments.fr_priamide(mol)
if pri_amide_groups > 0:
return "Y"
else:
return "N"
def AmineGroup(smi):
mol = Chem.MolFromSmiles(smi)
amine_groups = 0
amine_groups += Chem.Fragments.fr_Ar_NH(mol)
amine_groups += Chem.Fragments.fr_NH2(mol)
amine_groups += Chem.Fragments.fr_NH1(mol)
amine_groups += Chem.Fragments.fr_NH0(mol)
amine_groups -= Chem.Fragments.fr_amide(mol)
amine_groups -= Chem.Fragments.fr_amide(mol) #not an amide
amine_groups -= Chem.Fragments.fr_nitrile(mol) #not a nitrile
amine_groups -= Chem.Fragments.fr_Imine(mol) #not an imine
if amine_groups > 0:
return "Y"
else:
return "N"
def PrimaryAmineGroup(smi):
mol = Chem.MolFromSmiles(smi)
pri_amine_groups = 0
pri_amine_groups += Chem.Fragments.fr_NH2(mol)
if pri_amine_groups > 0:
return "Y"
else:
return "N"
def SecAmineGroup(smi):
mol = Chem.MolFromSmiles(smi)
sec_amine_groups = 0
sec_amine_groups += Chem.Fragments.fr_NH1(mol)
if sec_amine_groups > 0:
return "Y"
else:
return "N"
def TertAmineGroup(smi):
mol = Chem.MolFromSmiles(smi)
tert_amine_groups = 0
tert_amine_groups += Chem.Fragments.fr_NH0(mol)
if tert_amine_groups > 0:
return "Y"
else:
return "N"
def HetArAmineGroup(smi):
mol = Chem.MolFromSmiles(smi)
hetaramine_groups = 0
hetaramine_groups += Chem.Fragments.fr_Ar_N(mol)
hetaramine_groups += Chem.Fragments.fr_Nhpyrrole(mol)
hetaramine_groups += Chem.Fragments.fr_imidazole(mol)
hetaramine_groups += Chem.Fragments.fr_oxazole(mol)
hetaramine_groups += Chem.Fragments.fr_pyridine(mol)
hetaramine_groups += Chem.Fragments.fr_tetrazole(mol)
hetaramine_groups += Chem.Fragments.fr_thiazole(mol)
#more groups should go here - need to check - e.g. ??
if hetaramine_groups > 0:
return "Y"
else:
return "N"
def AnilineGroup(smi):
mol = Chem.MolFromSmiles(smi)
aniline_groups = 0
aniline_groups += Chem.Fragments.fr_aniline(mol)
if aniline_groups > 0:
return "Y"
else:
return "N"
def BasicNGroup(smi):
mol = Chem.MolFromSmiles(smi)
basic_n_groups = 0
basic_n_groups += Chem.Fragments.fr_piperdine(mol)
basic_n_groups += Chem.Fragments.fr_piperzine(mol)
basic_n_groups += Chem.Fragments.fr_oxime(mol)
basic_n_groups += Chem.Fragments.fr_morpholine(mol)
basic_n_groups += Chem.Fragments.fr_amidine(mol)
basic_n_groups += Chem.Fragments.fr_Imine(mol)
basic_n_groups += Chem.Fragments.fr_guanido(mol)
basic_n_groups += int(any([AmineGroup(smi)=="Y",
PrimaryAmineGroup(smi)=="Y",
SecAmineGroup(smi)=="Y",
TertAmineGroup(smi)=="Y",
HetArAmineGroup(smi)=="Y",
AnilineGroup(smi)=="Y"]))
if basic_n_groups > 0:
return "Y"
else:
return "N"
def EsterGroup(smi):
mol = Chem.MolFromSmiles(smi)
ester_groups = 0
ester_groups += Chem.Fragments.fr_ester(mol) # this also hits anhydrides
anhydride_smarts = '[CX3](=[OX1])[OX2][CX3](=[OX1])'
pattern = Chem.MolFromSmarts(anhydride_smarts)
matches = mol.GetSubstructMatches(pattern)
ester_groups -= 2*len(matches) #remove number of anhydride groups - two esters per anhydride
if ester_groups > 0:
return "Y"
else:
return "N"
def EtherGroup(smi):
ether_smarts = '[OD2]([#6])[#6]' # this also hits esters
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(ether_smarts)
matches = mol.GetSubstructMatches(pattern)
ether_groups = 0
ether_groups += len(matches)
ether_groups -= Chem.Fragments.fr_ester(mol) # not an ester
if ether_groups > 0:
return "Y"
else:
return "N"
def KetAlGroup(smi):
mol = Chem.MolFromSmiles(smi)
ketal_groups = 0
ketal_groups += Chem.Fragments.fr_ketone_Topliss(mol)
ketal_groups += Chem.Fragments.fr_ketone(mol)
ketal_groups += Chem.Fragments.fr_aldehyde(mol)
if ketal_groups > 0:
return "Y"
else:
return "N"
def CarbonylGroup(smi):
mol = Chem.MolFromSmiles(smi)
co_groups = 0
co_groups += Chem.Fragments.fr_C_O_noCOO(mol)
if co_groups > 0:
return "Y"
else:
return "N"
def HalogenGroup(smi):
mol = Chem.MolFromSmiles(smi)
halogen_groups = 0
halogen_groups += Chem.Fragments.fr_alkyl_halide(mol)
halogen_groups += Chem.Fragments.fr_halogen(mol)
if halogen_groups > 0:
return "Y"
else:
return "N"
def ChloroGroup(smi):
if 'Cl' in smi:
return "Y"
else:
return "N"
def BromoGroup(smi):
if 'Br' in smi:
return "Y"
else:
return "N"
def FluoroGroup(smi):
if 'F' in smi:
return "Y"
else:
return "N"
def IodoGroup(smi):
if 'I' in smi:
return "Y"
else:
return "N"
def SiliconeGroup(smi):
if 'Si'in smi:
return "Y"
else:
return "N"
def PhosporusGroup(smi):
if 'P' in smi:
return "Y"
else:
return "N"
def SulfurGroup(smi):
if (('S' in smi) and ('Si' not in smi)):
return "Y"
else:
return "N"
def HydrocarbonGroup(smi):
mol = Chem.MolFromSmiles(smi)
hydrocarb = 0
hydrocarb += Chem.Fragments.fr_unbrch_alkane(mol)
if hydrocarb > 0:
return "Y"
else:
return "N"
def NitrileGroup(smi):
mol = Chem.MolFromSmiles(smi)
nitrile = 0
nitrile += Chem.Fragments.fr_nitrile(mol)
if nitrile > 0:
return "Y"
else:
return "N"
def NitroGroup(smi):
mol = Chem.MolFromSmiles(smi)
nitro = 0
nitro += Chem.Fragments.fr_nitro(mol)
if nitro > 0:
return "Y"
else:
return "N"
def SulfideGroup(smi):
mol = Chem.MolFromSmiles(smi)
sulfide = 0
sulfide += Chem.Fragments.fr_sulfide(mol)
if sulfide > 0:
return "Y"
else:
return "N"
def SulfoneGroup(smi):
mol = Chem.MolFromSmiles(smi)
sulfone = 0
sulfone += Chem.Fragments.fr_sulfone(mol)
if sulfone > 0:
return "Y"
else:
return "N"
def UreaGroup(smi):
mol = Chem.MolFromSmiles(smi)
urea = 0
urea += Chem.Fragments.fr_urea(mol)
if urea > 0:
return "Y"
else:
return "N"
#define new functions that use SMARTS
def AlkeneGroup(smi):
alkene_smarts = 'C=C'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(alkene_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def AlkyneGroup(smi):
alkyne_smarts = '[CX2]#[CX2]'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(alkyne_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def AnhydrideGroup(smi):
anhydride_smarts = '[CX3](=[OX1])[OX2][CX3](=[OX1])'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(anhydride_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def SecAmideGroup(smi):
sec_amide_smarts = '[CX3][NX3;H1][CX3](=[OX1])[#6]'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(sec_amide_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def TertAmideGroup(smi):
tert_amide_smarts = '[NX3](-[CX4])(-[CX4])-[CX3]=[O]'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(tert_amide_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def AromaticGroup(smi):
'''This is very challenging. My approach is to get the rings, and see if any are aromatic.'''
# To detect aromatic rings, I would loop over the bonds in each ring and
# flag the ring as aromatic if all bonds are aromatic:
# https://www.rdkit.org/docs/Cookbook.html#identify-aromatic-rings
def isRingAromatic(mol, bondRing):
for id in bondRing:
if not mol.GetBondWithIdx(id).GetIsAromatic():
return False
return True
mol = Chem.MolFromSmiles(smi)
rings = mol.GetRingInfo()
aromatics = [isRingAromatic(mol, ring) for ring in rings.BondRings()] #returns boolean
if any(aromatics):
return 'Y'
else:
return 'N'
def CarbonateGroup(smi):
carbonate_smarts = '[CX3](=[O])(-[O])-[O]'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(carbonate_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def SulfoxideGroup(smi):
sulfoxide_smarts = '[S;D3](=[O])(-[C])-[C]'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(sulfoxide_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'
def SulfiteGroup(smi):
sulfite_smarts = '[S;D2](-[C])-[C]'
mol = Chem.MolFromSmiles(smi)
pattern = Chem.MolFromSmarts(sulfite_smarts)
matches = mol.GetSubstructMatches(pattern)
if len(matches) > 0:
return 'Y'
else:
return 'N'